مرض فقر الدم المنجلي
- 01 أكتوبر 2022
إعداد: أ. ريم العنزي
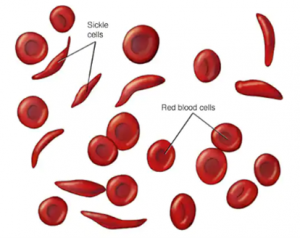
مرض فقر الدم المنجلي هو أحد الأنماط الصبغية المتنحية. والتي لا تحدث الإصابة بها إلاّ بعد أن يرث الشخص طفرتين وراثيّتين واحدة من الام والأخرى من الأب. ويؤثّر هذا المرض بشكل أساسي على نسبة كبيرة من خلايا الدم الحمراء المسؤولة عن نقل الأكسجين لخلايا الجسم. فيتسبب في حدوث تشوّه في شكلها بتحويله إلى ما يشبه الهلال أو المنجل (كما في الصورة الظاهرة). هذه الخلايا، عادة ما تتميز بشكلها الكروي الذي يمنحها مرونة عالية للحركة عبر الأوعية الدموية، وحدوث مثل هذا التشوه يؤثر بشكل كبير على مرونتها، فيجعلها جامدة ويبطّئ من حركتها عبر الأوعية الدموية، كما يمكن أن تعلق وتتراكم لتسبب انسداد في أحد هذه الأوعية. كما يتسبب ذلك أيضا بموت الخلايا خلال فترة قصيرة، ففي العادة تعيش خلايا الدم الحمراء إلى نحو 120 يوما، لكن مع تشوه شكلها، تقل فترة صلاحيتها إلى ما بين (10-20) يوم فقط [1]، وهو ما يتسبب في ظهور اعراض فقر الدم (الأنيميا)، نتيجة نقص أعداد خلايا الدم الحمراء الطبيعية التي تحمل الأكسجين لخلايا الجسم.[2]
بعض المؤشرات والإحصاءات المرتبطة بفقر الدم المنجلي
بالرغم من عدم وجود إحصائيات عالمية موثوقة عن مدى انتشار هذا المرض [3]، تشير بعض التقارير والدراسات إلى أنّه يصيب ملايين الأشخاص حول العالم، خاصّة في دول إفريقيا الصحراوية، والهند وعدد من دول حوض البحر الأبيض المتوسط، وكذلك المملكة العربية السعودية. كما تشير بعض الدراسات إلى وجود ارتباط وثيق ما بين انتشار فقر الدم المنجلي ومناطق انتشار الملاريا. وبناءا على هذه الدراسات فإنّ الإصابه به تحد من احتمالية العدوى بالملاريا بنسبة 90%.
يبلغ معدل الانتشار العالمي لمرض فقر الدم المنجلي نحو 112 شخص لكل 100000 شخص، بمعنى (0.11%) من إجمالي عدد سكان العالم بناءا على إحدى الدراسات التي قامت بتحليل بيانات 67 دراسة في عدة دول من العالم. لكن على الرغم من ذلك يُلاحظ وجود اختلافات كبيرة في معدل الانتشار بين تلك الدول، وخاصّة ما بين الدول المتقدمة وبعض الدول النامية. فعلى سبيل المثال يبلغ هذا المعدل 10 أضعاف المعدل العالمي في بعض الدول الإفريقية، فيما يقل عن نصف المعدل العالمي في دول أخرى من أوروبا. كما تشير الدراسات والتقارير إلى ارتباط مرض فقر الدم المنجلي ببعض الأعراق البشرية، وخاصة الأفارقة ذوي البشرة السوداء. فبناءا على بعض الإحصائات، وُجد أنه هناك مصاب واحد من كل 365 مواطن أمريكي من أصل إفريقي، وتزيد هذه النسبة بشكل كبير لمن يحملون سمة فقر الدم المنجلي، والتي تبلغ 1 إلى 13. [4،5]
بعض الإحصاءات من المملكة العربية السعودية
وفقا لموقع وزارة الصحة السعودي، يبلغ معدل الانتشار في عموم مناطق المملكة حوالي 0.26%، وهو معدّل أعلى من المعدل العالمي (0.11%) المذكور أعلاه. فيما بلغت نسبة حاملي سمة فقر الدم المنجلي حوالي 4.2% من إجمالي عدد السكان. ومن الملاحظ أرتفاع هذه المعدّلات بشكل كبير في المنطقة الشرقية، حيث يبلغ معدل الإصابة حوالي 1.2%، أما معدل انتشار سمة المرض فيبلغ حوالي 17% (بناءا على إحصائيات عام 2019). [1]
أسباب حدوث المرض
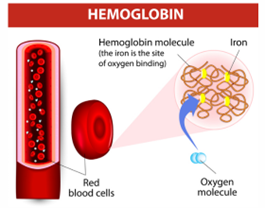
يتكون الهيموغلوبين من 4 وحدات فرعية من البروتين، وهي اثنتان (ألفا-غلوبين)، واثنتان (بيتا-غلوبين). ويسمى الجين الذي يسبب المرض (الجين الذي حدثت فيه طفرة جينية) هيموغلوبين الوحدة الفرعية بيتا (HBB)، وهو الجين المسؤول عن انتاج الوحدات البنائية (بيتا-غلوبين). تحدث لهذا الجين طفرات مختلفة تؤثر كل منها على نوع وحدات (بيتا-غلوبين) التي يتم إنتاجها. فعلى سبيل المثال، يتم بناءا على طفرة ما إنتاج نوع من وحدات الهيموغلوبين يسمى (هيموغلوبين S ) بدلا من (بيتا-غلوبين)، وبناءا على طفرات أخرى إنتاج (هيموغلوبين C)، أو (هيموغلوبين E) بدلا من (بيتا-غلوبين).
في حالة فقر الدم المنجلي يتم استبدال واحدة على الأقل من وحدات (بيتا –غلوبين) الاثنتين الطبيعيتين ب(هيموغلوبين S). لكن في الغالب يتم استبدال كلا الوحدتان ب (هيموغلوبين S)، وتسمى هذه الحالة فقر الدم المنجلي المتماثل (homozygous). [6]
أعراض المرض
تبدأ أولى أعراض المرض بعد (4-5) أشهر من الولادة، وعادة ما تختلف أعراض ومضاعفات هذا المرض بين شخص وآخر، فتظهر لدى البعض أعراض خفيفة، فيما تكون شديدة لدى آخرين. ومن الملاحظ عادة أنّ الحالة المرضية لفقر الدم تزداد سوءا كلّما مرّ الوقت، وعادة ما تُستخدم الأدوية المختلفة لتقليص احتمالية حدوث المضاعفات وبالتالي إطالة فترة حياة الأشخاص المصابين، أو لتخفيف الألم. [2]
تظهر على مصابي فقر الدم المنجلي أعراض عديدة ومختلفة أهمها الأنيميا، والعدوى المتكررة، واعتلال الطحال، والنخر اللاوعائي (موت أنسجة العظام) نتيجة ضعف وصول الدم، وارتفاع تعداد خلايا الدم البيضاء، وعدوى العظام، وارتفاع عدد الصفائح الدموية أو الخلايا الحمراء غير الناضجة، بالإضافة إلى متلازمة اليد والقدم، وأزمات الألم، واللتان تحدثان نتيجة انسداد الأوعية الدموية وبالتالي حدوث ألم شديد في منطقة الإصابة، وغيرها العديد من الأعراض الأخرى. [2،7]
العلاج
حتى الآن لا يوجد علاج جذري للعديد من المصابين بهذا المرض، – على الرغم من وجود بعض العلاجات الجراحية التي تحمل درجة خطورة عالية كزراعة الخلايا الجذعية للأطفال والمراهقين-. وبالتالي فإن معظم العلاجات التي تشمل أدوية مختلفة ووحدات الدم تهدف لتخفيف الألم أو لعلاج المضاعفات. ومن الأدوية التي يتم وصفها لمرضى فقر الدم المنجلي (Hydroxyurea)، و(Endari) الذان يعملان على تخفيف الألم وتقليص الحاجة لوحدات الدم، بالإضافة إلى حقن (Adakveo)، وغيرها من أصناف الأدوية الأخرى. [8]
الوقاية من المرض
لسوء الحظ لا يمكن (على الأقل حتى الآن) أن تتم وقاية الأشخاص الذين وُلدوا بجين معيب (طفرة جينية) من هذا المرض. لكن هناك بعض الإجراءات التي تتخذها العديد من الدول لتقليص فرص انتقال المرض لدى الأجيال اللاحقة. فعلى سبيل المثال في المملكة العربية السعودية وبعض الدول الأخرى، يتم إجراء فحوصات شاملة للمقبلين على الزواج لتقليل احتمالية انتقال مثل هذه الأمراض. [9،10]
كيف يمكن أن يساعد الفحص الجيني الآباء الحاملين لسمة فقر الدم المنجلي لإنجاب أطفال أصحاء؟
على الرغم من أنها طريقة معقدة نوعا ما، إلّا أنها ليست مستحيلة، ويتم الاعتماد عليها من قبل بعض الأشخاص حول العالم. وتتم هذه الطريقة عبر تقنية الإخصاب في المختبر (IVF)، حيث يتم إخصاب بويضة الأم مع الحيوانات المنوية للأب في المختبر، لتنمو البويضة المخصبة في ظروف المختبر حتى تتكون الكيسة الأريمية (blastocyst) بعد حوالي (5-6) أيام، ليتم بعد ذلك سحب عينة منها وإجراء الفحص الجيني المسمى PGT-M، وتحديد إيجابية أو سلبية الإصابة بمرض فقر الدم المنجلي ليُترك الخيار بزراعة ال(blastocyst) في الرحم أم لا للوالدين. [11]
______________
المراجع: